
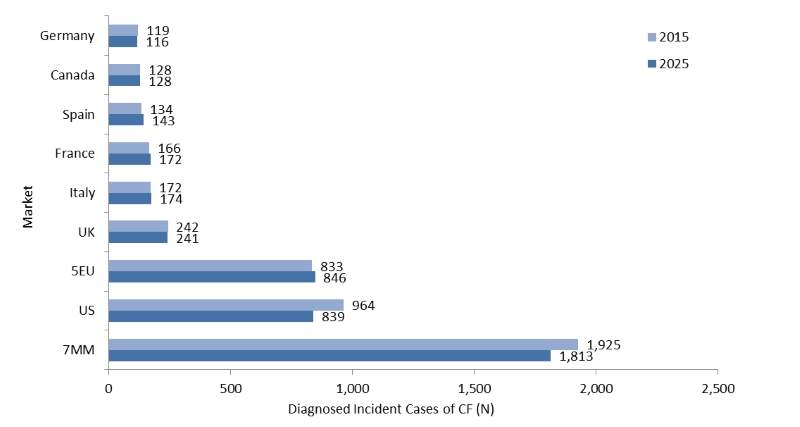
The diagnosed incident cases of cystic fibrosis (CF) are expected to decrease between 2015 and 2025 in the seven major markets (7MM) of the US, France, Germany, Italy, Spain, the UK, and Canada.
GlobalData epidemiologists forecast a decrease in the number of diagnosed cases, most notably in the US. Figure 1 presents the expected changes in the 7MM from 2015 to 2025 for all ages.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
In the 7MM, GlobalData epidemiologists forecast that the total diagnosed incident cases of CF will decrease from 1,925 cases in 2015 to 1,813 cases in 2025, at an annual growth rate (AGR) of -0.58%. The US will have the highest number of diagnosed incident cases of CF among the 7MM throughout the forecast period, while Germany will have the lowest.
Figure 1: 7MM, Diagnosed Incident Cases of CF, All Ages, Both Sexes, N, 2015 and 2025 (Forecast Based on Registry Data)

Source: GlobalData

US Tariffs are shifting - will you react or anticipate?
Don’t let policy changes catch you off guard. Stay proactive with real-time data and expert analysis.
By GlobalDataThe decreasing trend in incidence rates can be attributed to improved screening techniques that are now more readily available. Newborn CF screening has also been recommended by clinicians and CF groups as an early means of identifying asymptomatic patients so as to initiate early therapy to prevent long-term impacts of the disease. If a family member has CF or is a CF carrier, their offspring have an increased chance of also being CF carriers, and so carrier testing information can be helpful to people who may be thinking of having children.
CF is an inherited disorder that affects the cells that produce mucus, sweat, and digestive juices. This causes severe damage to the lungs, digestive system, and other organs. These secreted fluids are normally thin and slippery, but in people with CF, mutations in the CFTR gene cause the secretions to become sticky and thick. Instead of acting as a lubricant, the secretions plug up tubes, ducts, and passageways. In the lungs, the mucus clogs the airways and traps bacteria.
In the pancreas, the mucus prevents the release of digestive enzymes that allow the body to break down food and absorb vital nutrients. People with CF can have a variety of symptoms, including sweat with high salt content, persistent coughing, frequent lung infections (including pneumonia or bronchitis), wheezing or shortness of breath, poor growth or weight gain despite having a good appetite, frequent stools that are greasy and bulky or difficulty with bowel movements, and male infertility.
Details about the trend analysis and other discussions of CF epidemiology can be found in the EpiCast Report: Cystic Fibrosis – Epidemiology Forecast to 2025.
