
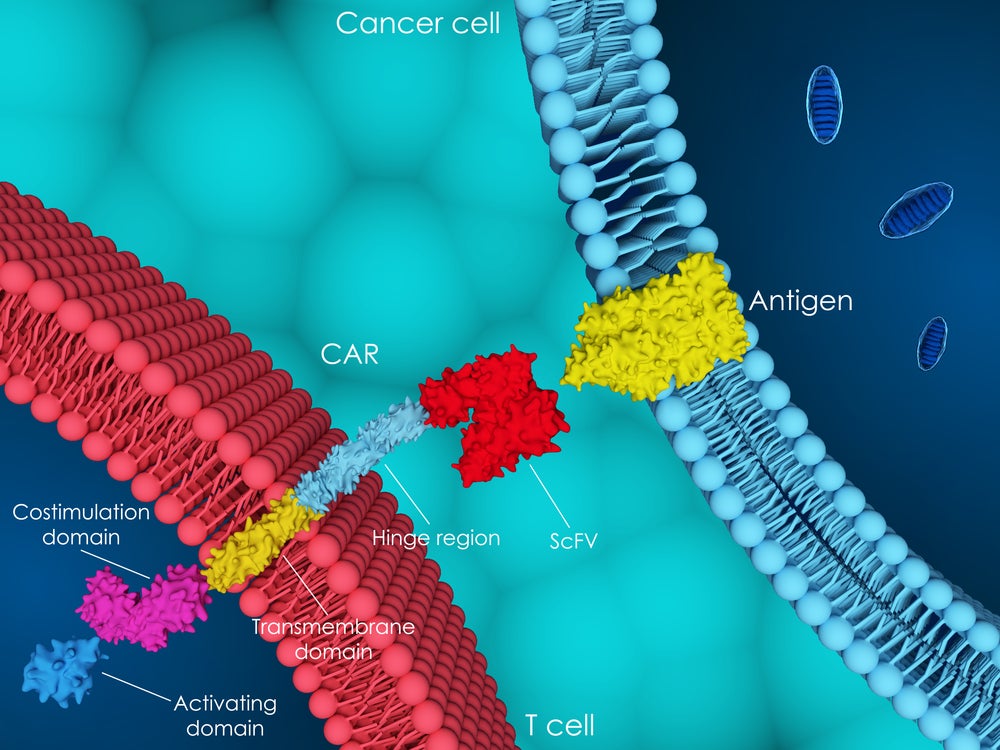
Chimeric antigen receptor (CAR) T-cell therapies have drastically changed treatment paradigms in relapsed and refractory (R/R) haematological malignancies since the approval of the first CAR-T, Novartis’ Kymriah (tisagenlecleucel) in 2017. Since then, the US Food and Drug Administration (FDA) has approved six more CAR-T cell therapies for several indications, but there are several barriers to the uptake and commercialisation of these novel assets.
These therapies require primary T cells to be collected from the patient, be modified ex vivo, undergo expansion and quality control, and be re-introduced. This process may take between two and seven weeks – during which disease could worsen. Due to this long vein-to-vein time, patients also require bridging therapy, typically a toxic chemotherapy regimen, which can add complications. Before being dosed with the modified T cells, the patient undergoes a course of lymphodepletion to improve cell engraftment, increasing the risk of infection, which can be fatal for those with advanced haematological cancers. There are also serious safety concerns due to a high risk of cytokine release syndrome (CRS) or immune effector cell-associated neurotoxicity syndrome (ICANS). The complex production of the CAR-Ts and the management of the side effects require complex infrastructure, resulting in limited patient access and high cost compared to other therapeutic options.
Go deeper with GlobalData

Despite these issues for CAR-Ts, there is still a high interest in the modality and investment in overcoming these issues. One idea being investigated is in vivo CAR-T cell therapies. These function by dosing a patient with a gene therapy vector that targets their T cells with a payload that will express the CAR protein. The vectors and payloads come in many forms – the most developed being lentiviral vectors with DNA payloads that integrate into the host genome.
In vivo therapies sidestep many ex vivo drawbacks
Alternative methods include lipid-based nanoparticles as the vector and mRNA as the payload, but these are limited by low transduction efficiency and transient expression, respectively. In vivo CAR-T cell therapies sidestep many of the drawbacks of ex vivo CAR-Ts, such as the long vein-to-vein time. Patients can be dosed quickly with an off-the-shelf vector, eliminating the need for bridging therapies and specialised treatment centres. In vivo CAR-Ts also avoid the requirement of lymphodepletion before dosing, as they rely on the host’s T cells to function. All these advantages are likely to result in a significantly lower cost.
One concern with in vivo CAR-Ts is the potential insertional mutagenesis of the lentiviral DNA payload, causing secondary cancers. Ex vivo CAR-Ts already carry an FDA-mandated warning about potential T-cell malignancy due to insertional mutagenesis. However, quality control before dosing could lower this risk. With in vivo engineered T cells, no quality check before dosing is possible, especially for off-target tissue transduction. We will need to wait for the clinical safety data, as well as to learn if there are reductions in CRS and ICANS.
Umoja BioPharma and EsoBiotec are the two leaders in the in vivo CAR-T cell therapy space. AstraZeneca recently announced its intention to acquire EsoBiotec for up to $1 billion. Novartis entered the field by partnering with Vyriad in November 2024 to combine its cell therapy expertise with Vyriad’s in vivo lentiviral-targeted delivery platform. Big pharma’s involvement with this novel modality at such an early stage demonstrates optimism about in vivo CAR-T’s potential. In addition, according to GlobalData’s proprietary drug database, there are currently 12 in vivo CAR-T assets in the preclinical and 14 more in the discovery stage of development for haematological malignancies and solid tumours.

US Tariffs are shifting - will you react or anticipate?
Don’t let policy changes catch you off guard. Stay proactive with real-time data and expert analysis.
By GlobalDataAstraZeneca and Novartis are getting a head start
EsoBiotec’s lead asset is ESO-T01, a lentiviral vector encoding a BCMA-targeting CAR, which is in a Phase I trial in R/R multiple myeloma (MM) and dosed its first patient in January 2025. ESO-T01 would be competing with existing CAR-Ts in R/R MM, such as BMS’ Abecma (idecabtagene vicleucel) and Johnson & Johnson’s Carvykti (ciltacabtagene autoleucel). Umoja has two assets in Phase I clinical trials: UB-VV400, a lentivirus carrying a CD22 CAR payload, which is in a trial in R/R diffuse large B cell lymphoma and UB-VV111, a lentivirus carrying a CD19 CAR payload, in a trial in R/R chronic lymphocytic leukaemia and other B-cell malignancies.
These indications have also previously approved ex vivo CAR-Ts. However, the risk for Umoja and EsoBiotec comes not from the marketed CAR-Ts but from the second generation of ex vivo CAR-Ts in late-stage clinical development, such as Galapagos’ GLPG5101, an ex vivo CAR-T in the Phase II ATALANTA-1 trial in R/R non-Hodgkin lymphoma. Preliminary results from ATALANTA-1 were positive, and decentralised manufacturing implemented by Galapagos solves many of the issues with existing CAR-Ts, such as the long vein-to-vein time and the requirement for specialised treatment centres. When Umoja and EsoBiotec assets reach the clinic, they will compete with new agents that would have already displaced the existing ex vivo CAR-Ts.
AstraZeneca and Novartis are getting a head start on early in vivo CAR-T development to hedge their bets in the highly competitive cell therapy market segment. GlobalData’s proprietary likelihood of approval (LoA) algorithm gives ESO-T01 a 22% LoA in R/R MM compared to an indication benchmark of 11%. Likewise, UB-VV400 has a 17% LoA versus an indication benchmark of 8%, and UB-VV111 has a 19% LoA compared to an indication benchmark of 12%.
CAR-Ts will play a pivotal role in blood cancer treatment in the future, but whether they are in vivo or ex vivo remains to be seen.
